
1.Science:第一次发布完整的人类基因组序列
doi:10.1126/science.abj6987
由于当时全世界称赞的人类基因组测序工作不完整,当时DNA人类基因组能读取人类基因组的某些部分。即使更新后,它仍然缺乏大约8%的人类基因组。从端粒到端粒的新研究(Telomere-to-Telomere, T2T)该联盟的研究人员描述了历史上第一个完整的人类基因组——一套建造和维持人类的指令——测序。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是The complete sequence of a human genome”。

华盛顿大学研究员Evan Eichler一些使我们独特的人类基因实际上是在基因组的暗物质中完全遗漏的。花了20多年的时间,但我们终于完成了。Eichler参与当前的研究工作和最初的人类基因组计划(Human Genome Project)。
科学家指出,人类基因组的全貌将使人们更多地了解人类的进化和生物学特征,也为衰老、神经退行性疾病、癌症和心脏病等领域的医学发现打开大门。参考资料:
Sergey Nurk et al. The complete sequence of a human genome. Science, 2022, doi:10.1126/science.abj6987.
2.Science:完整的基因组和组和表观遗传图谱
doi:10.1126/science.abl4178
纺锤丝为了在细胞分裂过程中忠实地将遗传物质分配给子细胞,必须通过一种方式称为动粒(kinetochore)”的结构与DNA结合,每个染色体中都有动粒的丝粒(centromere)上组装。人类的丝粒位于一串中被称为α重复卫星序列(alpha satellite, αSat)的串联重复DNA在序列中,这些序列通常色体上数百万个碱基对。一大串αSat序列通常被其他类型的串联卫星重复序列包围,包括可转录的基因。由于其规模和重复性,以前的基因组测序工作无法完全组装富含卫星重复序列的区域,从而限制了研究其分布、变异和功能的能力。
在过去的20年里,人类参考基因组中几乎没有丝粒周边区和丝粒(pericentromeric and centromeric, peri/centromeric)卫星DNA序列。从端粒到端粒的新研究(Telomere-to-Telomere, T2T)联盟的研究人员使用完整的端粒到端粒(T2T)为了揭示这些卫星重复序列在大长度和小长度尺度上的分化和进化模式,开发并部署了定制的计算方法。他们还做了一些准确绘制的实验αSat重复序列与动粒蛋白相互作用。最后,他们比较了多个个体之间的比较peri/centromeric了解这些序列在不同的遗传背景下是如何变化的。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是Complete genomic and epigenetic maps of human centromeres”。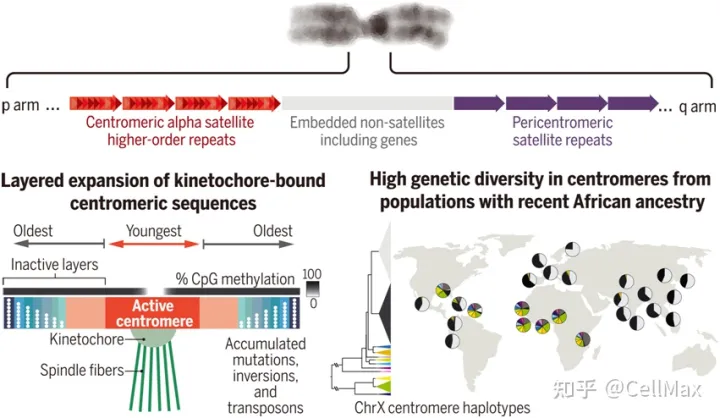
重复序列占用卫星T2T-CHM13基因组的6.2%,其中αSat它是最大的组成部分(占基因组的2.8%)。对每一颗丝粒αSat对序列关系的详细研究表明,全基因组的证据表明,人类着丝粒是通过分层扩张进行的(layered expansions)”进化的。具体来说,每个着丝粒区域出现不同的重复序列变异,并通过类似的连续串联重复(tandem duplication)随着时间的推移,老侧翼序列缩小和分化。
这些作者还发现,每串作者也发现αSat最近内部扩张的重复序列更有可能与内部动粒蛋白-丝粒蛋白A(CENP-A)—相互作用,这与CpG减少甲基化的区域是一致的。这表明局部卫星重复序列扩张、动粒定位和DNA低甲基化关系密切。此外,他们还发现了影响各种卫星重复序列类型的大型和意想不到的结构重排,包括活跃的丝粒αSat。参考资料:
Nicolas Altemose et al. Complete genomic and epigenetic maps of human centromeres. Science, 2022, doi:10.1126/science.abl4178.
3.Science:完整的人类参考基因组改进了对人类遗传变异的分析
doi:10.1126/science.abl3533
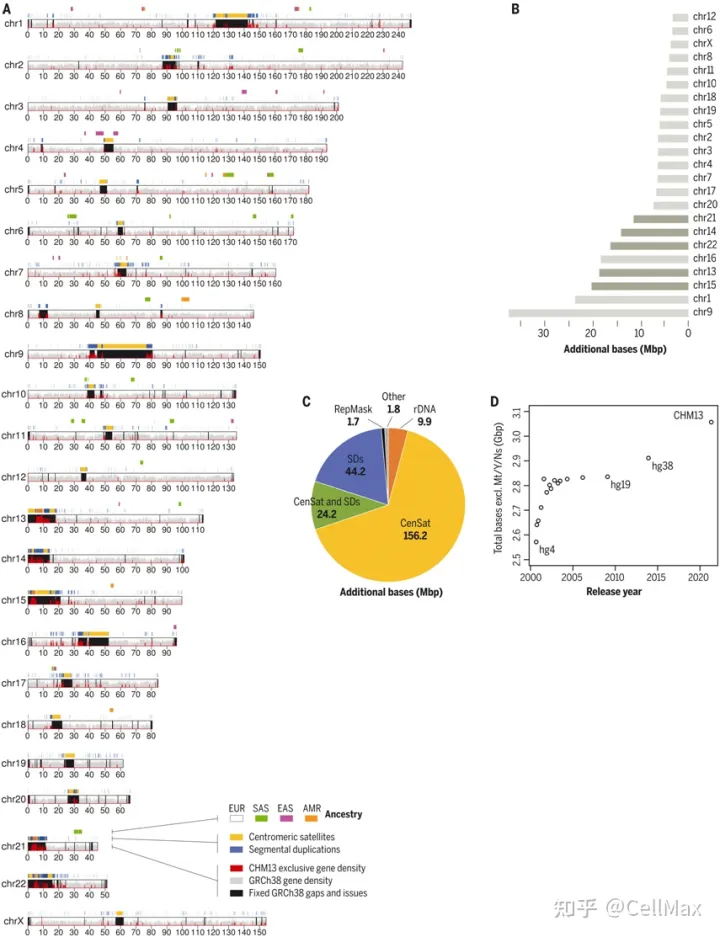
作为几乎所有人类基因组研究的比较基线,人类参考基因组的核心应用之一。不幸的是,人类参考基因组的许多困难地区几十年来一直没有得到解决,并受到塌陷区、序列缺失等问题的影响。与目前的人类参考基因组相比,GRCh38,人类T2T-CHM13(Telomer-to-Telomere CHM13)基因组填补了所有剩余空白,增加了近200个基因组序列 Mbp(Mbp),校正了成千上万的结构错误,释放了人类基因组中最复杂的区域进行科学探索。
从端粒到端粒的新研究(Telomere-to-Telomere, T2T)联盟研究人员展示T2T-CHM参考基因组是如何在全球不同的队列中普遍改进序列映射和变异识别的。这个队列包括扩大的1000基因组计划(1)KGP)所有3202个样本经过短读测序,全球17个不同样本经过长读测序。用最先进的方法调用单核苷酸变异(SNV)和结构变异(SV),他们记录了T2T-CHM与以往人类参考基因组序列的优势和局限性相比,13强调有望在基因组技术挑战区揭示新的生物学见解。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是A complete reference genome improves analysis of human genetic variation”。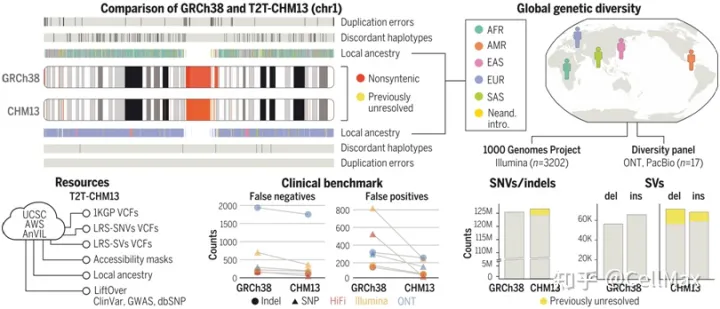
在1KGP样本中,与GRCh与38相比,这些作者在全基因组范围内使用T2T-CHM13发现了100多万个额外的高质量变异。在以前未解决的基因组区域,他们在每个样本中发现了数十万个变异——这是进化和生物医学发现的好机会。T2T-CHM13提高了602个三人组之间的孟德尔一致性,消除了每个样本数以万计的假性SNV,假阳性率降低了269个具有挑战性的医学相关基因的12倍。这些校正在很大程度上是因为GRCh由错误坍塌区或重复区引起的38中长达9 Mbp70个蛋白编码基因在上述不准确序列中的改进。通过使用T2T-CHM参考基因组还可以更好地了解整个基因组范围内的结构变异,大大提高序列插入与缺失之间的平衡。最后,通过T2T-CHM13提供大量资源(包括1)KGP基因类型、可访问性掩码和突出的注释数据库),他们的研究将从当前的人类参考基因组推广到T2T-CHM13过渡。
参考资料:
Sergey Aganezov et al. A complete reference genome improves analysis of human genetic variation. Science, 2022, doi:10.1126/science.abl3533.
4.Science:揭示完整人类基因组重复序列的转录和表观遗传状态
doi:10.1126/science.abk3112
从端粒到端粒的新研究(Telomere-to-Telomere, T2T)联盟的研究人员实施了一个全面的重复序列注释工作流程,使用以前已知的人类重复序列和重复序列建模,然后手动组织,包括基因注释评估重叠、分段复制、串联重复序列和注释重复序列。通过这种方法,他们开发了一个更新的人类重复序列目录,并复序列注释。他们在T2T-CHM在13发现了43个以前未知的重复序列和重复序列变异(repeat variant),经常携带基因的携带基因的19个复杂的复合重复序列结构。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是From telomere to telomere: The transcriptional and epigenetic state of human repeat elements”。
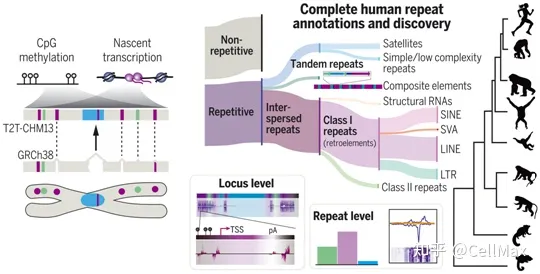
精确使用PRO-seq(precision nuclear run-on sequencing)牛津纳米孔技术公司生成的技术和长读测序数据CpG这些作者在全基因组范围内对逆转录因子进行了评估RNA聚合酶的结合揭示了新生转录、序列分歧、CpG密度与甲基化的相关性。他们将这些分析扩展到评估所有重复序列RNA聚合酶占有率,包括在所有人类染色体之前无法进入的高密度卫星重复序列。
此外,在早期发育阶段和完整的细胞周期时间序列中,这些作者使用依赖图谱和不依赖图谱的方法来发现整个卫星重复序列RNA聚合酶占有率很低;相反,转座因子转录丰富,作为CpG甲基化与丝粒亚结构变化的界限。这些数据共同揭示了活跃的逆转录因素和DNA新的重复序列家族和复合重复序列的衍生和进化潜在机制是甲基化之间的动态关系。
关注新兴HG002 X这些作者揭示了人类群体可能存在高水平的重复序列变异,包括影响基因复制数的复合重复序列复制数。此外,他们强调了重复序列对基因组结构多样性的影响,揭示了人类和灵长类动物之间重复序列的扩展,并对逆转录因导事件进行了高度可信的注释。参考资料:
Mitchell R. Vollger et al. From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science, 2022, doi:10.1126/science.abk3112.
5.Science:揭示完整人类基因组中的片段重复及其变异
doi:10.1126/science.abj6965
大、高同一度的重复序列-称为片段重复(segmental duplication, SD)—它通常是基因组中最被测序和组装的区域。虽然人类参考基因组是建立的SD景观提供了路线图,但基因组剩余的50%以上的空白对应于复杂的空白SD区域。SD它是进化基因创新的主要来源,在类人猿物种内部和之间的遗传变异中起着不成比例的作用。人类基因组完整:T2T-CHM科学家可以识别基因,发现人类的遗传变异模式。
从端粒到端粒的新研究(Telomere-to-Telomere, T2T)联盟的研究人员在T2T-CHM13中发现了51 Mbp(million base pair,额外的人类碱基对数百万)SD,现在估计7%的人类基因组是由人类基因组估计的SD在人类基因组中,总共有31亿个碱基对,SD共有218 Mbp。SD占短臂近端着丝的三分之二(68.1Mbp中的45.1Mbp),这些SD它是人类基因组中最大的(见图中的A部分)。SD占短臂近端着丝的三分之二(68.1Mbp中的45.1Mbp),这些SD它是人类基因组中最大的(见图中的A部分)。此外,54%的近端有丝绸SD复制数是可变的,或者在被研究的六个人中映射到不同的染色体上。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是Segmental duplications and their variation in a complete human genome”。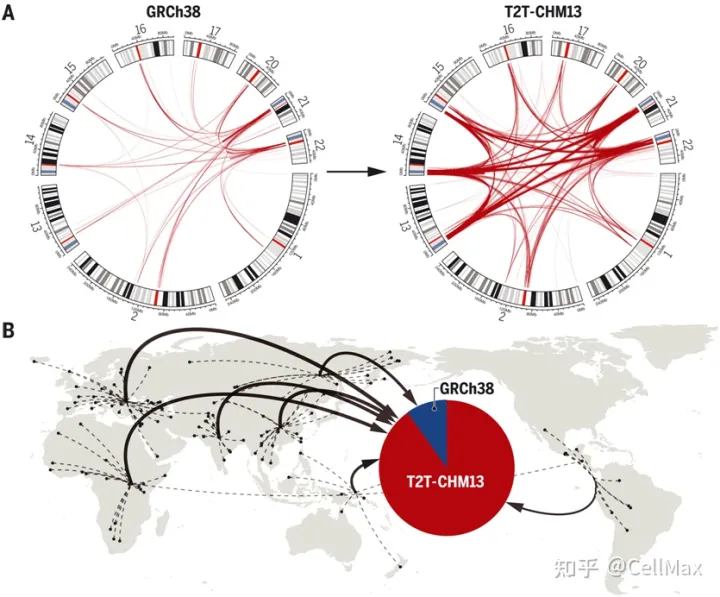
目前参考基因组(GRCh38)和T2T-CHM13的SD详细比较含量,发现有81Mbp以前未解决或结构可变的SD。短读全基因组序列数据来自一个由268人组成的多样性小组,显示人类拷贝数和T2T-CHM13匹配的可能性是GRCh38匹配9倍(前者为59.26 Mbp,后者为6.55Mbp),包括119个蛋白编码基因(见图中B部分)。
这些作者调查了人类遗传变异的模式,发现结构和单核苷酸变异的模式,发现结构和单核苷酸多样性显著增加。TBC1D3)这些区域在个体之间的差异达到数十万个碱基对和基因拷贝,显示出一些最高的全基因组结构杂合度(85%至90%)。参考资料:
Mitchell R. Vollger et al. Segmental duplications and their variation in a complete human genome. Science, 2022, doi:10.1126/science.abj6965.
6.Science:人类基因组表观遗传模式的完整分析
doi:10.1126/science.abj5089
人类参考基因组已成为许多大规模计划的基础,包括表观基因组(epigenome)表观基因组是控制基因活动和细胞功能的一组标记,以及蛋白质的相互作用。然而,在过去的20年里,建立一个完整的表观基因组的努力一直受到不完整的人类参考基因组的阻碍。
随着最近技术的进步,从端粒到端粒(Telomere-to-Telomere, T2T)联盟的研究人员现在可以通过完整的人类基因组端粒组装到端粒:T2T-CHM综合研究人类基因组的结构和功能。因此,人们现在可以扩大人类表观基因组,包括2.25亿碱基对的额外序列。2022年4月1日发表了相关研究结果Science在期刊上,论文的标题是Epigenetic patterns in a complete human genome”。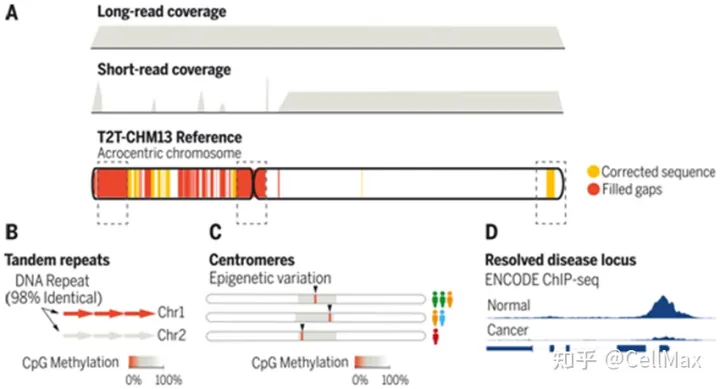
表观基因组是指DNA修饰(如CpG甲基化),蛋白质-DNA它们共同影响基因表达、基因组调节和基因组稳定性。这些表观遗传特征在细胞分裂时是可遗传的,但在发育过程中是动态的,产生了不同组织和细胞类型的独特模式。在这项新研究中,这些作者提出了人类基因组的表观遗传注释,如探索未解决的区域,包括近端染色体短臂和片段重复基因(segmentally duplicated genes)以及各种重复序列类别,包括人类着丝粒。完整的表观遗传%的人类基因组进行完整的表观遗传注释,为阐明这些基因组件的功能提供了基础,这对人们了解基因组的调节、功能和进化至关重要。
参考资料:
Ariel Gershman et al. Epigenetic patterns in a complete human genome. Science, 2022, doi:10.1126/science.abj5089.
7.Science:青铜时代和铁器时代的人口流动是新疆人口历史的基础
doi:10.1126/science.abk1534
与山脉接壤的中国新疆是一个重要的历史区域。在一项新的研究中,我们的科学家对古代基因组进行了取样,研究了从青铜时代(约5000年至3000年)到铁时代(约3000年至2000年)的人口变化,再到历史时代(约2000年)。这一分析表明,年长的个体代表了草原文化的祖先,东亚和中亚的祖先在青铜时代末期和铁器时代初期进入该地区。在历史时期,混合继续进行,但核心草原成分保留,使人口形成遗传连续体。在中心人群中保持这种遗传连续性是令人惊讶的,因为它代表了孤立人群中更典型的观察模式。此外,这些遗传联系确定了以前未知的谱系,可以解释印欧语系的传播。
8.Science:关键基因是实验性食品网络持续存在的基础
doi:10.1126/science.abf2232
在过去的几十年里,关键物种的识别,即关键物种的识别生态系统中发挥重要作用的物种,在不同的系统中增加。Barbour人们将这一概念扩展到基因上,表明特定植物防御基因的单等位基因促进了整个小型实验营养系统的物种共存。具体来说,这种等位基因的植物生长得更快,支持两种食草动物及其捕食者的更大种群。这一发现表明,基因变异可以在有机系统的结构和功能中发挥作用。
9.Science:探讨哺乳动物的脑化率
doi:10.1126/science.abl5584
哺乳动物物中,哺乳动物的大脑与身体大小的比例最大。人们一直认为,这种关系出现在哺乳动物进化的早期,扩大的大脑引导它们进入一种新的、多样化的形式。然而,Bertrand等人研究了古新世以来所有哺乳动物的脑化率,发现身体大小首先增加,以便在恐龙灭绝后进行生态填充。后来,大脑的大小开始增加,这可能是因为在日益复杂的环境中需要更大的认知能力。这导致了包括人类大脑在内的今天高度脑化的大脑。
10.Science:脊髓小胶质细胞参与缓解和复发性神经痛
doi:10.1126/science.abf6805
对神经系统的损害从病理上改变了身体感觉系统,这可能导致神经性疼痛。虽然疼痛已经得到了很好的研究,但协调疼痛恢复的机制仍然不清楚。Kohno等人在神经损伤的小鼠中发现了一只CD11c+疼痛发生后出现脊髓小胶质细胞,对神经病理性疼痛的恢复至关重要。这些细胞促进疼痛恢复的能力取决于它们的高水平胰岛素样生长因子-1(IGF1)的表达。即使疼痛恢复后,CD11c+小胶质细胞仍然存在,如果性就会恢复。这些发现阐明了缓解和复发性神经痛的机制,并可能有助于发展治疗策略。
11.Science:选择性社会学习择性社会学习保留了复杂的认知算法
doi:10.1126/science.abn0915
我们世代积累复杂算法的能力,使人类能够适应不同的环境,解决超出我们个人限制的挑战。然而,很难解释创新算法的文化积累。Thompson为了探索不同学习条件下算法的演变,等人研究了大量的参与者。选择性社会学习涉及不同策略或模型的成功水平,比随机社会学习或尝试的非社会学习更能保存难以发明的高效算法。许多人使用两种有效的算法,但最有效的算法只在选择性的社会学习下传播。

